Getting started with moffragmentor#
Installation#
We recommend installing moffragmentor in a clean virtual environment (e.g., a conda environment)
You can install the latest stable release from PyPi using
pip install moffragmentor
or the latest development version using
pip install git+https://github.com/kjappelbaum/moffragmentor.git
Note
If you install via pip you will need to manually install openbabel (e.g. conda install openbabel -c conda-forge).
If you want to determine RCSR codes, you will also need to install a Java runtime environment (JRE) of version 1.5.0 or later on your machine as we use the Systre (Symmetry, Structure (Recognition) and Refinement) code to perform analysis of the labeled quotient graphs we construct in moffragmentor.
Extensions#
In case you want to use the show_molecule(),
or show_structure() function in Jupyter lab you have to
jupyter-labextension install @jupyter-widgets/jupyterlab-manager
jupyter-labextension install nglview-js-widgets
jupyter-nbextension enable nglview --py --sys-prefix
You also might find the debugging help in the nglview documentation useful.
Fragmenting a MOF#
To fragment a MOF you need to create an instance of MOF
and then call fragment().
from moffragmentor import MOF
mof = MOF.from_cif(<my_cif.cif>)
fragmentation_result = mof.fragment()
The result is a FragmentationResult namedtuple with the fields nodes, linkers,
both subclasses of a moffragmentor.sbu.SBUCollection and bound_solvent, unbound_solvent, both moffragmentor.molecule.NonSbuMoleculeCollection, and a moffragmentor.net.Net.
Warning
If you use the MOF.from_cif method, we will run pymatgen.analysis.spacegroup.SpacegroupAnalyzer on the input structure.
This might take some time, and we also have encountered cases where it can be really slow.
If you do not want this, you can either “manually” call the constructor or tune the tolerance parameters.
Warning
Note that moffragmentor currently does not automatically delete bound solvent. This is due to two observations:
We have very little understanding of what solvent we can remove without affecting the structural integrity.
We (currently) do not have a way to estimate if a solvent is charged. We explore different implementation strategies, but we do not have a robust one at this moment.
You might want a quick overview of the composition of the different components.
You can access this via the composition properties
solvent_collection.composition
which will return a dictionary of the counts of the compositions, for example {'C3 H7 N1 O1': 3, 'H2 O1': 4}.
Clearly, we do not consider floating solvent for the computation of the net.
Known issues
For some structures in the CSD MOF subset, there will be problems with the fragmentation.
One example is CAYSIE, which is a metalloporphyrinate.
Here, the code struggles to distinguish nodes and linkers
as a core routine of the moffragmentor is to check if a metal atom is inside another, potential linker, molecule.
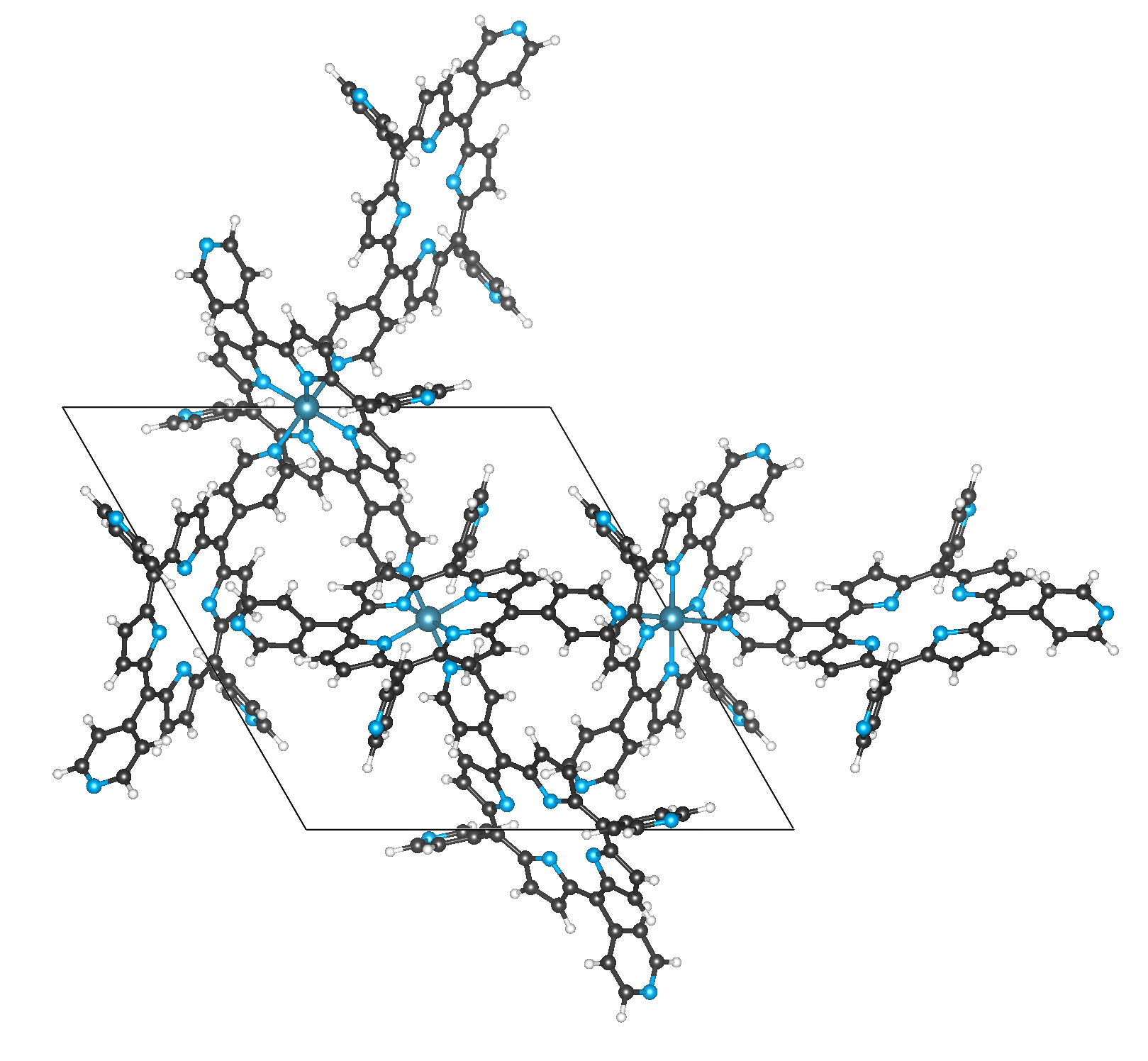
Fig. 1 Example of a metalloporphyrinate for which the fragmentor fails.#
Also note that there are problems with analyzing the combinatorial topology of 1D rod MOFs. There only recently has been an algorithm proposed that is implemented in ToposPro.
Customizing the log level
moffragmentor uses the loguru for logging. By default, logging from moffragmentor is disabled to not interfere with your logs.
However, you can easily customize the logging:
import sys
from loguru import logger
# enable moffragmentor logging
logger.enable("moffragmentor")
# define the logging level
LEVEL = "INFO || DEBUG || WARNING || etc."
# set the handler
# for logging to stdout
logger.add(sys.stdout, level=LEVEL)
# or for logging to a file
logger.add("my_log_file.log", level=LEVEL, enqueue=True)
In many cases, however, you might find it convenient to simply call enable_logging()
from moffragmentor.utils import enable_logging
enable_logging()
which will enable logging with sane defaults (i.e. logging to stderr for INFO and WARNING levels).